复旦大学附属眼耳鼻喉科医院舒易来教授团队详解遗传性耳聋基因治疗的进展、前景及挑战
- 2023-03-04 08:00:21 健康一线
- 健康
超过全球人口的5%,即4.66亿人患有致残性听力障碍(中度以上听力障碍),其中3400万是儿童。中国致残性听力障碍患者约7000万,每年新生3万聋儿,其中60%是由遗传因素也就是基因缺陷引起。尽管目前已有150多种耳聋基因得到鉴定,但临床上尚无可以治疗的药物。随着生物医药技术的革新和发展,基因治疗目前被认为是根治遗传疾病最有前景的策略。基因治疗(gene therapy)是一种通过操纵目的基因来实现疾病治疗的技术,主要包括基因替代(gene replacement)、基因抑制(gene suppression)和基因编辑(gene editing)等策略。
近日,复旦大学附属眼耳鼻喉科医院舒易来教授团队在美国细胞与基因治疗学会会刊 Molecular Therapy上发表了题为:Advances in gene therapy hold promise for treating hereditary hearing loss 的综述论文。
该论文系统、全面论述了遗传性耳聋的分类及细胞学机制,并重点阐述了三种主要的基因治疗策略在遗传性耳聋临床前试验中的成功应用及现有临床试验进展,最后讨论了遗传性耳聋基因治疗的现有挑战和未来应用前景。

Molecular Therapy是国际上基因和细胞治疗领域顶级期刊,该杂志国际审稿人对于该论文给予了高度评价,认为“本文主要通讯作者舒易来医生作为具有强大科学背景的临床医生,在耳聋基因治疗领域做出了多项重要的原创性贡献”,同时评价“本文对临床转化的总结和观点会引起读者兴趣、富有意义”。("The senior authors have been active in the field for a number of years and haveauthored several important original articles…Written by clinicians with a strong scientific background, the translational aspects of the research are highlighted and an overview of recently approved clinical trials is presented together …Overall, I believe that the article is of interest to the readers of Molecular Therapy …").
就遗传性耳聋而言,鉴于内耳是一个相对封闭且充满液体(内淋巴液和外淋巴液)的腔室,理论上,基因治疗药物(如病毒、核苷酸、核蛋白复合体等)通过局部给药后可随淋巴液扩散至众多内耳靶细胞而相对不易大量外泄,因此内耳比较适合开展基因治疗。
随着基因操控工具的不断迭代更新,尤其是CRISPR-Cas9系统的出现和不断优化,近年来遗传性耳聋的基因治疗取得了令人瞩目的进展。在临床前研究中,超过40项研究利用基因治疗策略成功纠正了20余个致聋基因相关动物模型的听力。值得一提的是,全球已有三项遗传性耳聋的基因治疗临床试验被批准,其中包括复旦大学附属眼耳鼻喉科医院的舒易来教授团队主导的项目,该项目率先完成了全球首例OTOF遗传性耳聋患者的体内给药,该领域我国学者走在了国际最前沿。
1.遗传性耳聋的分类
根据除听觉系统外是否有其它器官的临床体育bd,遗传性耳聋可分为综合征型听力障碍(syndromic hearing loss,SHL)和非综合征型听力障碍(non-syndromic hearing loss,NSHL)。
1.1.非综合征型遗传性耳聋
约70%的遗传性耳聋患者为非综合征型。NSHL最常见的遗传方式是常染色体隐性遗传(75%-80%),其次是常染色体显性遗传(20%),X染色体连锁遗传(<2%)和线粒体遗传(<1%)。迄今为止,已有120多个基因被报道与NSHL相关。其中GJB2是最常见的致病基因(21.6%),其次是STRC(16.1%)、SLC26A4(6.6%)和TECTA(5.2%)。
1)常染色体隐性NSHL(ARNSHL)通常是语前聋,常表现为所有频率的重度到极重度耳聋。到目前为止,已有78个基因突变被报道与ADNSHL相关,包括GJB2、STRC、SLC26A4、GJB6、TMC1、OTOF、CDH23、MYO6和SYNE4等。
2)常染色体显性NSHL(ADNSHL)通常是语后聋,其耳聋程度比ARNSHL轻。已有51个耳聋基因被报道,常包括编码电压门控钾离子通道的KCNQ4基因、编码α-tectorin的TECTA基因、WFS1基因和COCH基因的突变。有趣的是,某些ADNSHL在不同频率下有不同程度的听力损失,例如,KCNQ4基因突变通常导致高频听力损失,而WFS1基因突变患者则导致低频听力损失。
3)X染色体连锁和线粒体非综合征型耳聋NSHL相对少见。5个基因(POU3F4、PRPS1、SMPX、AIFM1和COL4A6)与X连锁NSHL相关,其中转录因子POU3F4的突变是最常见的致聋原因。线粒体12S核糖体核糖核酸(rRNA)的A1555G突变和C1494T突变是线粒体遗传模式,不同于直接引起的听力损失,这两个线粒体基因的突变会导致患者对氨基糖苷类抗生素非常敏感,从而在给予治疗剂量的药物时会发生听力损失。
1.2.综合征型听力损失
SHL常伴有其它系统的临床表现,包括眼睛、心脏、肾脏、神经系统、皮肤、骨骼等,占遗传性耳聋的30%。SHL中以Pendred综合征、Usher综合征(USH)和Waardenburg综合征(WS)最为著名;而USH、Pendred综合征和Jervell and Lange-Nielsen综合征(JLNS)已在临床前动物模型研究中成功获得内耳基因治疗。
1)Usher综合征以感音神经性耳聋、视网膜色素变性和前庭功能障碍等为主要临床特征。在40种影响视力的听力损失中,高达50%是由Usher综合征引起,每10万人有3.5 ~ 6.2人患病。目前,USH有两种分类方式,包括临床异质性和遗传异质性分类。就临床异质性而言,根据发病年龄、表型严重程度和前庭功能是否受影响,USH可细分为三种临床分类:USH1、USH2和USH3。就遗传异质性而言,每种亚型的USH可归因于多个基因突变——USH1(MYO7A、USH1C、CDH23、PCDH15、USH1G和CIB2)、USH2(USH2A、ADGRV1和WHRN)和USH3(CLRN1)。这些基因编码的蛋白质在内耳耳蜗和前庭以及视网膜的光感受器细胞中表达,因此解释了为什么USH会同时影响听觉和视觉系统。
2)Pendred综合征也称为耳聋-甲状腺综合征,是最常见的常染色体隐性SHL之一,患病率约为7.5/100,000。患该综合征的病人不仅表现于导水管扩张,而且还可表现出前庭功能障碍以及甲状腺肿。目前,已确定Pendred的致病基因有:SLC26A4、FOXI1和KCNJ10。大约一半以上的病例与SLC26A4突变相关。SLC26A4基因编码pendrin蛋白,是一种多功能阴离子交换蛋白,主要表达于内耳和甲状腺,对耳蜗内淋巴和甲状腺细胞的离子稳态至关重要。
3)Jervell and Lange-Nielsen综合征(JLNS)表现为心律失常和感音神经性耳聋,患病率为1.6-6 / 1,000,000。该类患者心律失常的主要表现为QT间期延长。目前认为JLNS与两个耳聋基因(KCNQ1和KCNE1)突变有关,它们分别导致JLSN1和JLSN2,其中KCNQ1突变较为常见(约占90%)。这两个致病基因编码的离子通道蛋白对心脏和耳蜗的功能至关重要。
4)Waardenburg综合征(WS)是一种听觉-色素性疾病,常表现于常染色体显性遗传,患病率为1/42,000。研究发现,常导致该疾病的基因主要包括EDN3、EDNRB、MITF、PAX3、SNAI2和SOX10等。鉴于这些基因对黑素细胞的形成和发育至关重要,因此,除听力损失外,WS以头发、皮肤和眼睛色素沉着丧失为特征。对此,根据突变基因所伴随的体育bd,将WS分为4种亚型。其中,1型和2型较常见,3型和4型较罕见。
2.遗传性耳聋的细胞学机制
哺乳动物的耳蜗由不同类型的细胞组成,这些细胞因其独特的基因表达模式而具有不同的功能。根据基因在耳蜗不同组织或细胞中的表达分布,耳聋基因大致分为三大类(图1):
1) 在听觉毛细胞(HCs)中表达的基因;
2) 在支持细胞(SCs)中表达的基因;
3) 在血管纹(SV)中表达的基因等。
已有的研究显示,这三类致病基因是内耳基因治疗的常见靶点。当然,也有部分耳聋基因表达于耳蜗其它部位,如螺旋神经节神经元(SGN)、内外沟细胞、盖膜和Reissner膜等。通常,这些致病基因的表达部位为病毒载体的选择和治疗窗口的评估以及基因治疗系统的设计提供了有力的指导。
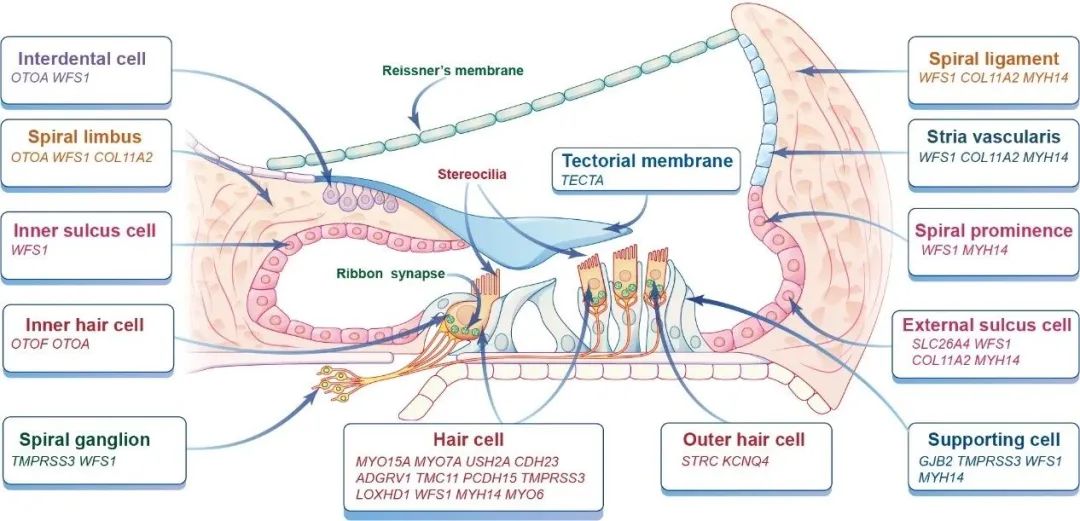
图1:遗传性耳聋常见致病基因 (诊断率≥1%) 的主要表达部位
2.1.毛细胞耳聋基因
超过一半的耳聋基因在耳蜗毛细胞中表达,因此毛细胞更受到基因治疗的关注。耳蜗内有两种类型的毛细胞:对声音做出反应的单排内毛细胞(inner hair cells,IHCs)和放大声音的三排外毛细胞(outer hair cells,OHCs)。在HCs的顶端表面有一束用于机械电转导的静纤毛;在HCs的基底有负责将神经递质释放到SGN的带状突触。常见主要表达于内外毛细胞的基因包括MYO15A、MYO7A、USH2A、CDH23、MYO6、TMC1、PCDH15等。然而,另外一些基因则主要表达于IHCs或OHCs。例如,对带状突触的神经传递至关重要的OTOF和对传入突触的谷氨酸释放至关重要的VGLUT3主要在IHCs中表达,而对毛束凝聚至关重要的STRC、对钾离子稳态至关重要的KCNQ4和对细胞核定位至关重要的SYNE4基因主要在OHCs中表达。
2.2.支持细胞耳聋基因
具有支持功能的SCs对维持HCs的结构完整和功能正常具有重要意义。GJB2、GJB3和GJB6主要表达在SCs中,分别编码缝隙连接蛋白26、31和30。在NSHL病例中,GJB2和GJB6的变异占比大,而GJB3的突变较罕见。缝隙连接蛋白26和30是耳蜗中两个主要连接蛋白,在SCs中组装成缝隙连接,对于K+、Ca2+以及细胞信号和营养分子交换至关重要。
2.3.血管纹耳聋基因
血管纹位于耳蜗外侧壁,在维持内淋巴内高浓度K+的过程中起关键作用,而内淋巴内高浓度K+是HC发挥功能的先决条件。SV由三层细胞(边缘细胞、中间细胞和基底细胞)组成,通过离子通道将钾离子从外淋巴泵到内淋巴。因此,与离子通道(如KCNQ1、KCNE1和KCNJ10)或细胞层完整性所需的紧密连接分子(如CLDN11)相关的基因突变可导致听力障碍。
3.遗传性耳聋的基因治疗
根据遗传性疾病的遗传类型和致病机制,不同的致病机制的基因突变应选用合适的基因治疗策略(图2)。隐性遗传病主要是由两个等位基因的功能缺失(loss-of-function,LOF)引起;显性遗传病更为复杂,有三种主要的分子机制:1)单倍剂量不足(haploinsufficiency,LOF),指的是一个等位基因的功能缺失,但另一个等位基因不足以维持基因的功能;2)显性负(dominant-negative,DN)效应,即突变等位基因不仅无功能,而且干扰正常等位基因产物的功能;3)功能获得(gain of function,GOF),指的是致病性等位基因基因产物剂量或活性增加(图2B)。
不同的基因治疗策略有着不同的工作方式(图2A)。基因替代策略是通过递送正常基因以提供充足的有功能蛋白,适用于隐性遗传病和单倍剂量不足的显性遗传病(图2)。基因抑制策略是通过反义寡核苷酸(ASO)或RNA干扰(RNAi)技术在mRNA水平抑制突变基因表达,该方法可用于DN和GOF导致的显性遗传性疾病(图2)。基因编辑策略主要包括CRISPR/Cas9核酸酶非同源末端连接(NHEJ)修复方式介导的基因敲除(gene disruption)策略,通过敲除突变基因,可应用于DN和GOF导致的显性遗传性疾病;以及CRISPR/Cas9介导的同源重组修复(HDR)、碱基编辑器和先导编辑器介导的基因修复(gene correction)策略,可直接纠正基因突变位点,因此显性、隐性遗传性疾病均适用(图2)。
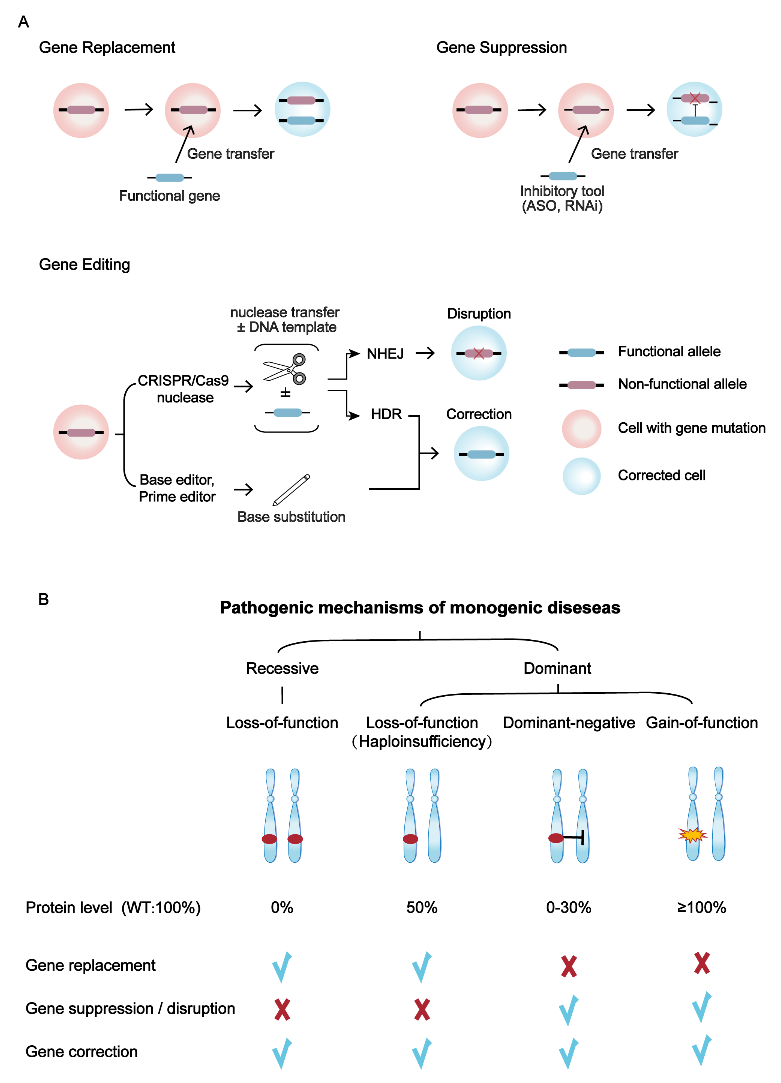
图2 单基因疾病的基因治疗策略。(A) 三种主要的基因治疗策略。(B)单基因疾病的发病机制及对应的基因治疗方法。
3.1.基因替代
基因替代治疗是最常见的基因疗法,已被广泛用于治疗隐性遗传性耳聋。自2012年在Vglut3敲除小鼠模型中报道了首例有效的遗传性耳聋基因替代治疗研究以来,大量遗传性耳聋的基因替代治疗研究相继涌现。其中研究较多的基因替代治疗靶点包括TMC1、USH相关基因、OTOF等。针对这些靶点,递送顺利与否也成了基因治疗成功的关键。
当前,尽管AAV是内耳基因替代治疗最常用的生物递送工具,但由于其有限的包装容量(~4.7 kb),很难实现大基因的递送。令学界欣喜的是,经过科学家们不懈的努力,终于开发出基于核酸重组或蛋白重组方法的双AAV递送策略,以解决较大耳聋基因的递送问题。其中也包括舒易来教授团队开发的蛋白水平重组技术,该团队采用AAV-PHP.eB成功地将OTOF基因约6 kb的cDNA分为两部分共同递送至Otof-/-小鼠内耳中,并重组成完整的OTOF蛋白,成功恢复了该小鼠模型的听力,回复到接近完全正常。
由于基因替代疗法具有效率高和操作易等特性,其在治疗遗传性耳聋方面取得了很大的成功。然而,该策略也存在一些局限。例如,由于此方法无法根据细胞的需要准确地调控基因的表达,因此可能存在基因过度表达或异位表达的风险。此外,由于单个AAV的负载能力有限,通过多个病毒载体介导的大外源基因递送仍具有挑战性。
3.2.基因抑制
基因抑制疗法可以在不干扰DNA的情况下在RNA水平进行调控从而改变基因的表达,主要包括两种技术:ASO和RNAi。
1)ASO是经过修饰的合成核酸序列,能通过结合互补的RNA序列上调/下调基因的表达,或改变蛋白质的异构体。ASO可作为基因抑制疗法的一种工具,通过降解靶mRNA或调节mRNA的选择性剪接来抑制突变基因的表达。ASO治疗策略主要被应用与USH1C基因的c.216G>A突变,该突变导致了阿卡迪亚人群中全部的I型Usher综合征。USH1C c.216G>A突变产生了一个隐式剪接位点,导致USH1C mRNA及其编码的harmonin蛋白被截短。因此,与基因替代策略相比,ASO是一种理想的阻断USH1C基因异常剪接位点的方法。然而,ASO目前仅被报道用于USH的体内治疗。舒易来团队最新研究发现ASO在体外耳聋患者细胞中,可有效纠正Slc26a4耳聋基因中国人最常见的c.919-2A>G突变的错误剪接。然而,ASO是否适用于其他类型的遗传性耳聋还需要进一步探索。
2)RNAi技术是指通过短干扰RNA(siRNA)和微小RNA(miRNA)中和靶向mRNA分子来抑制基因表达。siRNA以及miRNA已在动物模型中被分别应用于治疗GJB2 p.R75W以及TMC1 c.T1253A 突变引起的遗传性耳聋。除了这些传统的RNAi工具,CRISPR/Cas13 RNA编辑系统已成为一种具备更高特异性的新型RNA干扰工具。
最近,舒易来团队采用贝多芬小鼠(Tmc1基因突变小鼠)探索了Cas13d的潜在治疗作用。该团队发现CRISPR/Cas13系统在体内降低了70%的Tmc1突变mRNA,从而改善了小鼠的听觉功能。这项概念验证研究证明了RNA编辑工具治疗遗传性耳聋的安全性和有效性。然而,基于cas13的RNA编辑系统是否适用于成年小鼠遗传耳聋的治疗尚需要进一步研究。
基因抑制治疗策略在不导致永久DNA破坏的情况下沉默基因的表达,具有较大的应用前景。然而,该策略是否存在潜在的脱靶效应以及基因表达不完全/暂时阻断的潜在性仍有待探索。
3.3.基因编辑
基因编辑是通过碱基的添加、删除和替换目标位置改变DNA的序列。主要包括基因敲除和基因修复等策略。
1)基因敲除
2014年哈佛大学的著名基因编辑和听觉医学专家 David Liu、Zheng-Yi Chen、舒易来等合作,国际上首次报道CRISPR/Cas9在体内对内耳细胞成功进行了基因编辑(以SpCas9-gRNA核糖核蛋白复合体)。随后,David Liu 和 Zheng-Yi Chen 将SpCas9-gRNA技术应用于TMC1 Bth耳聋小鼠模型,改善了该小鼠的听觉功能。此后,基因敲除技术已被成功应用于MYO6、KCNQ4、PCDH15等基因的显性负效应突变。
针对Myo6 p.C442Y突变,舒易来团队开发了一个高效的靶向毛细胞的基因敲除治疗系统,将该系统注射到Myo6WT/ C442Y新生小鼠的内耳中可特异性敲除突变等位基因,从而改善突变小鼠的听力功能。然而,由于耳蜗组织中含有许多非靶向细胞,在体内很难准确评估基因敲除效率,而基因敲除效率是评估治疗效果和未来临床转化的关键因素之一。为了解决这一局限性,该团队以了Atoh1-GFP; Kcnq4 c.683G>A杂交两种突变小鼠模型,成功标记耳聋模型中耳蜗毛细胞,用于精确检测毛细胞内的基因编辑效率。该团队研究发现分选出标记的阳性细胞的编辑效率可达34.1%,而通过检测整个耳蜗的编辑效率仅为1.45%,该研究表明准确检测毛细胞中的编辑效率方法的成功建立,并证明Cas9核酸酶可以在体内高效编辑毛细胞。
2)基因修复
鉴于大多数遗传性耳聋是由隐性遗传性突变引起,这些突变需要被修复,而不是被敲除。因此,基因修复工具(包括碱基编辑器、先导编辑器和Cas9核酸酶介导的HDR)可以精确纠正突变位点,是很有前景的治疗方法。由于HDR的编辑效率低,碱基插入或缺失副产物水平高,且通常仅在细胞分裂时活跃,因此利用该方法治疗遗传性耳聋的研究难度很大。有团队报道,以CRISPR/Cas9介导的HDR在小鼠受精卵中纠正了Cdh23ahl突变等位基因,挽救了其相关的年龄相关听力损失。随后,舒易来等团队采用了HDR的优化版—同源介导的末端连接(HMEJ)在体修复了Klhl18纯合隐性突变小鼠模型的听觉功能,听力维持时间至少6个月。
随着基因修复工具的推陈出新,新型的基因编辑工具如碱基编辑器以及先导编辑器能够在不断裂DNA的前提下对基因进行直接的修复。目前主要的碱基编辑器有:催化C→T(或G→A)转换的胞嘧啶碱基编辑器(CBEs)和介导A→G(或T→C)转换的腺嘌呤碱基编辑器(ABEs)。由于人类的大多数遗传病是由点突变引起的,DNA编辑器在遗传病的治疗中具有巨大的潜力。David Liu团队已成功将CBE应用于隐性Tmc1突变的Baringo小鼠模型,该系统可使内耳中C•G点突变逆转为野生型T•A,从而改善小鼠的听力功能,但是因为编辑效率低等原因,听力只维持了大约2周。
碱基编辑器的局限性之一是太大而无法被包到单个AAV中。最近,David Liu团队通过最小化ABE和AAV的组分,成功设计了一种单AAV ABE系统,当在小鼠眼眶后递送时,该系统在许多器官(如肝脏、心脏和肌肉)显示出较高的碱基编辑效率。此外,未来可以通过提高载体的递送效率和碱基编辑器的表达来进一步推进这一策略。
除了DNA编辑器,舒易来联合杨辉教授团队开发出了既不切割DNA,也不切割RNA的RNA碱基编辑器治疗策略,可在RNA水平进行碱基的精确校正。该研究团队研究了基于Cas13的RNA ABE在Myo6C442Y/+小鼠的治疗效果,研究结果表明该RNA编辑系统能够纠正突变的Myo6等位基因,并恢复小鼠听觉功能,证明了RNA碱基编辑器在治疗遗传性耳聋中的重要价值。
然而,碱基编辑器目前尚不能实现所有12种碱基改变类型。一种名为“先导编辑器”的新型基因编辑工具克服了这一障碍,该工具可以纠正12种点突变类型,同时也能实现DNA片段的插入或缺失。目前,先导编辑器已在成年小鼠视网膜中成功编辑了Dnmt1基因,但其是否适用于遗传性耳聋的治疗尚未报道,值得进一步研究。
在内耳基因治疗的研究中,基因校正工具仍被认为是一种极具前景的基因治疗选择,但仍需要进一步提高其编辑效率等。
4.遗传性耳聋基因治疗临床试验
针对遗传性耳聋的基因治疗,已有包括我国舒易来团队项目在内的3项针对OTOF基因突变治疗的临床试验正在进行中。另外两个项目由美国两家医药公司——Akouos和Decibel Therapeutics实施。
Akouos公司设计的AK-OTOF试验性新药申请(IND)是美国食品药品监督管理局(FDA)于2022年9月批准的第一份遗传性耳聋基因治疗新药申请。AK-OTOF是由泛启动子控制,由AAVAnc80载体负载的人耳畸蛋白cDNA的候选产品,目的是在内耳中表达有功能的耳畸蛋白。该团队临床前研究表明, AK-OTOF促进了otoferlin蛋白在小鼠和灵长类动物内毛细胞中的表达,并恢复了Otof-/-小鼠长达至少6个月的听力功能。该候选药物的安全性已被证实,其并未导致小鼠和灵长类动物的耳蜗功能以及全身组织病理学异常。Akouos正计划在儿童中开展I/II期临床试验,以评估其在人体中的安全性、耐受性和有效性,前两名儿童参与者年龄在7岁左右,随后招募的参与者年龄可能最小为2岁。
随后,Decibel Therapeutics公司开发的DB-OTO于2022年10月获得了FDA的批准,该公司预计将在2023年上半年启动I/II期临床试验。DB-OTO的人otoferlin cDNA由双AAV1载体携带,并由毛细胞选择性启动子控制。其在小鼠进行的临床前研究表明,DB-OTO给药可使Otof-/-小鼠的听觉功能持续改善至52周龄。在非人灵长类动物的临床前研究表明,局部耳蜗内注射后otoferlin在耳蜗基底膜全长表达。该公司的临床试验人群与Akouos大致相同,但其后续将招募包括两岁以下的儿童。
舒易来团队联合上海鼎新基因科技有限公司研发的基因治疗候选药物RRG-003是由AAV载体携带的人otoferlin cDNA。该团队通过圆窗给药纠正了Otof-/-小鼠的听力损伤,且持续时长达至少6个月。在临床试验中,该团队采用微创内耳给药。其纳入第一批患者年龄为3-10岁,第二批患者年龄将小于3岁的患者,该团队目前已完成全球首例遗传性耳聋患者的体内给药,目前正在进行安全性和听力改善评估。
5. 遗传性耳聋基因治疗面临的挑战
5.1.成年鼠的干预
由于人的内耳在出生时就已经发育成熟,相当于已经成年的小鼠耳蜗,因此研究基因治疗策略能否成功挽救具有成熟内耳的成年小鼠模型的听觉功能具有重要意义。然而,在成年小鼠模型中进行基因治疗存在一些挑战。首先,在不干扰内耳淋巴环境的情况下,成年小鼠的内耳递送尤为困难。新生小鼠的耳囊是软骨的,可以用玻璃微管直接穿透。相比之下,成年小鼠的耳囊是完全骨化的,因此很难在不影响听觉功能的情况下进行骨性覆盖物注射。其次,病毒载体在成年小鼠内耳细胞中的转导效率较低,尤其是在外毛细胞、支持细胞中。因此,有必要进一步探索或优化具有更高成年小鼠耳蜗细胞转导效率的载体。
5.2.治疗效果的持续时间
由于哺乳动物内耳中的许多细胞被认为是终末分化并且缺乏再生能力的,因此内耳基因疗法的治疗在理论上是持久的。然而,在动物模型实践中,基因治疗的持续时间变化很大,从几周到几个月或一年不等。基因治疗的持续时间可能与特定基因导致的病理生理以及研究中使用的动物模型有关。此外,随着时间推移,基因治疗的疗效降低可能是因为产生了针对病毒载体和外源性基因的中和抗体,或者是由于基因治疗系统编辑效率低而未被纠正的内耳细胞本身的功能进行性恶化所致。因此,内耳基因治疗能否获得更持久、更稳定的治疗效果仍需进一步研究。从这个角度而言,基因编辑可能是一个较好的选择,因为基因编辑是永久性的,但其编辑效率仍需进一步提高。
5.3.递送载体的特异性、安全性和容量
许多耳聋相关基因仅表达在内耳的特定细胞中,需要一定的环境才能发挥作用,这意味着AAV转导的特异性具有重要意义。限制基因治疗系统在非靶向细胞中表达的一种方法是将细胞特异性miRNA结合元件与载体盒结合,从而用特异性miRNA沉默细胞中的表达。在靶细胞中调节基因表达的另一种方法是利用细胞特异性启动子。与CMV或CAG等泛启动子不同,针对特异细胞具有特异性AAV有其优势。但此领域仍然需要探索,尤其是那些足够小的启动子,以便包装于AAV载体中,同时为治疗元件留出足够的空间,以提高AAV载体的特异性。
对于AAV的安全性。一方面需要考虑机体对其衣壳的潜在免疫应答,尽管AAV的免疫原性通常低于其他病毒载体,通过对AAV衣壳进行改造或降低抗AAV抗体水平,可以规避AAV的免疫效应。
如前文所述,通过在内耳应用双AAV系统,已部分解决了AAV载体的容量限制。尽管这些研究获得了较好的结果,但全长基因的表达水平仍有待提高。这个问题可能是由于同一细胞被不同载体转导的概率较低,以及不同部分限速了基因重组过程。
5.4临床前试验动物模型
在内耳研究中,小鼠模型因其基因组特征明确以及与人类内耳非常相似而成为最常见的实验动物。然而,为了将基因治疗策略推进到人类临床试验,有必要在更大的动物模型(如非人灵长类动物)中测试有效性或安全性。已有研究通过成功地向恒河猴的内耳注射生理盐水,而不破坏内耳功能。基于这项研究,研究人员已经证明了AAV9-PHP.B、AAV-S、Anc80L65和AAV1在非人灵长类动物耳蜗内的有效转导。
6.遗传性耳聋基因治疗未来前景和展望
基因治疗是一种极具前景的遗传性耳聋治疗方法,它可以提供传统医学无法实现的听觉精准治疗。为进一步推进遗传性耳聋基因治疗的临床转化,未来的研究可以从以下几个方面进一步进行推进:
1)优化现有基因治疗工具的疗效和特异性,尝试应用先导编辑器等新兴工具提高治疗效果,同时最大限度地减少脱靶效应;
2)优化载体,提高其转导效率和特定细胞亚群的特异靶向性;
3)优化给药路径和方法,尽可能提高微创技术和水平,方便基因治疗系统安全递送到内耳;
4)由于CRISPR/Cas9系统的gRNA无法进入哺乳动物线粒体,因此可以重新利用目前已不常用但有潜力的基因编辑工具,如锌指核酸酶(ZFN)和转录激活因子样效应物核酸酶(TALEN)来治疗线粒体耳聋基因突变;
5)加强耳聋基因检测以诊断遗传性耳聋患者的遗传学病因,为临床医生提供更多关于特定突变和临床表型对应的信息,这对于确定内耳基因传递的合适治疗窗口和制定个性化治疗方案至关重要。
综上,内耳基因治疗的不断发展为遗传性耳聋的治疗提供了新的希望,并为其他遗传性疾病的治疗提供了新的借鉴。

本文地址://www.styjt.com/jiankang/2023-03-04/630411.html
友情提示:文章内容为作者个人观点,不代表本站立场且不构成任何建议,本站拥有对此声明的最终解释权。如果读者发现稿件侵权、失实、错误等问题,可联系我们处理

- 什么是蕈样肉芽肿2023-03-04 08:46:02
- 什么是轮状病毒腹泻2023-03-04 08:46:01
- 厌奶期是什么时候2023-03-04 08:46:00
- 喉咙难以下咽怎么回事2023-03-04 08:45:58
- 呼啦圈能减肥吗2023-03-04 08:45:57


 70岁以上老人核酸检测费用多少 新规定明天起不做核酸了吗
70岁以上老人核酸检测费用多少 新规定明天起不做核酸了吗2022-11-07

 梅婷现任70岁老公曾剑个人资料(曾剑个人资料)
梅婷现任70岁老公曾剑个人资料(曾剑个人资料)2022-09-08
- 烟台今天已封闭的小区 烟台现在封闭小区名单有哪些
2022-10-12 09:07:30
- 张家界桑植新娘吴梅婚纱照事件完整版 看女主出轨聊天内容视频
2022-11-11 14:27:00
- 目前南岸区及江北封闭小区名单有哪些 看最新封控通告
2022-11-11 14:49:26
- 无人售货机功能(无人自动售货机操作方法介绍)
2022-07-27 08:41:09
- 2022南宁封控小区名单表 南宁最新封闭小区名单2022年8月什么情况
2022-08-26 09:52:30


